
We hoped that sequencing the honey bee genome would unlock the evolutionary mysteries around the origin of insect societies. So twelve years later, how is it possible that we still only know what about two thirds of their genes actually do?
We used to think that knowing an organism’s genome sequence – or complete genetic code – meant that we could uncover all its biological secrets. Finally, we could understand species’ evolutionary origins. Knowing its genome, we could pinpoint what causes complex diseases or heritable behaviors. We could piece together metabolic pathways, helping to understand things like how nutrition influences health. Maybe we could even learn how to design a genome ourselves, from scratch.
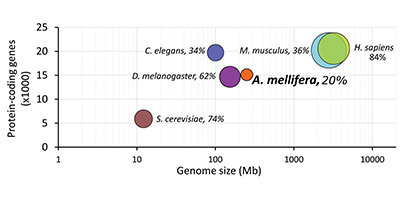
When I was an undergraduate student first starting to research honey bees, I quickly learned that is not actually the case. Our lab specializes in a technique called “proteomics,” which lets us take snapshots of the different protein molecules in extracts from crushed tissues, so we can learn things like what’s special about hygienic bees or what a virus infection does to the brain. Upon joining the lab, I found out that we (and everyone else in the world) routinely identify fewer proteins in honey bees than in other organisms, like mice or humans (Figure 1). It looked like up to a quarter of the honey bee data was just plain missing. How could this be? We have a genome and a database of genes and proteins, so there was no obvious reason why a honey bee sample should produce lower quality data than a mouse sample. And that sub-par quality meant that our research was suffering, since we were only seeing a fraction of the biological picture that we should. Trying to figure out the origin of this maddening deficiency sent me on a year-long quest to try to solve – or at the very least, understand – the problem. And it all starts with the genome.
Now I know that a genome can’t just be read like a book. It’s more like trying to read endless text written in a different language. Translating that information into useful knowledge that tells us how honey bees tick is a painstaking process. This is true for any genetic sequence, but the honey bee genome has been exceptionally troublesome, putting our best sequencing technology and de-coding tools to the test. The genome sequence has enabled astonishing advances in honey bee science (like the high-tech selective breeding program I wrote about previously1), but many times, we are still working half blind. Here’s why.
Flies First
The first insect genome to be sequenced was Drosophila melanogaster, or the common fruit fly. In fact, its genome was published back in the year 2000, even before the human genome in 2001. This might seem like a strange choice, but it dates back to the earliest genetic studies where male fruit fly mutants could be easily identified due to a change in eye color. This founded the fruit flies as extremely important model organisms, especially for genetics and neurobiology. Over the years, fruit fly research has born no less than six Nobel Prizes. The flies have been diligently studied for decades. MD Adams (lead author of the seminal Science paper describing the fruit fly genome sequence2) writes, “Drosophila melanogaster is one of the most intensively studied organisms in biology [emphasis my own] and serves as a model system for the investigation of many developmental and cellular processes common to higher eukaryotes, including humans.”
The fruit fly’s utility as a model system comes in large part because humans and flies, while having wildly different appearances, share around 60% of our DNA sequence. This means that many of our protein molecular machines are also the same, or very similar. Flies are also cheaper, faster, and easier to grow in the lab than mice or other mammals, so they quickly became a classic study. Because of this, by the time the genome was sequenced, we already had a wealth of knowledge about the fruit fly’s genes and what they do. When the complete genetic map was finally released, we knew exactly what to do with it: since we already knew what most of its genes looked like – the kinds of sequences they tended to have, and how they were arranged – it was relatively easy to write computer programs to find the rest of the genes we didn’t already know about.
No such luck with the honey bee.
Odd Genome Out
The trouble began during the sequencing process. Like any plant or animal, honey bees’ DNA is arranged into chromosomes – where humans have 23 chromosomes, honey bees have 16, which is pretty normal. So far so good. Each chromosome is made up of millions of molecular building blocks, which we call nucleotides, abbreviated as A, T, G, and C. Most genomes are made up of around 55-60% A and T nucleotides, with the rest being G and C. Fruit flies have 58% A and T, mosquitoes have 55%, and humans have 59%. Honey bees, however, are a clear outlier, with an average of 67% A and T. This might not seem like it should be a big deal, but it causes some serious problems.
To illustrate, imagine you’re reading an article. It’s a normal story, maybe something like you might find in this magazine. It’s fairly easy for you to read it from start to finish because it’s arranged in a way that makes sense: a variety of words are used, and they have a logical flow. You seldom lose your place or have to start again. Now imagine you’re reading an article with only a limited set of words (say, 10). Since many of the sentences would inevitably be the same, this repetition makes it easy to lose your place. Was I on line 3 or line 5? They both say the same thing, so I’m not sure . . .
This is a lot like what happens when we try to sequence a genome with long, AT-rich regions: they’re so repetitive, it’s hard to know what the right order is. This is at least part of the reason why the initial honey bee sequencing efforts in 2006 generated an incomplete genome, at around 88% coverage. That was still enough to start trying to unlock the genome’s secrets, but the havoc wreaked by its AT-richness wasn’t over.
Cracking the Code
An assembled genome has little utility on its own; first, it needs to be annotated, and the AT-richness impaired this procedure as well. Genome annotation is the process of finding and describing the thousands of genes that reside in the genome. It’s important because most modern molecular biology tools (including our lab’s specialty, proteomics) rely in some way on having a complete, accurate set of predicted genes or proteins to meaningfully interpret the ….